publications
our interesting findings 🕵️
2026
-
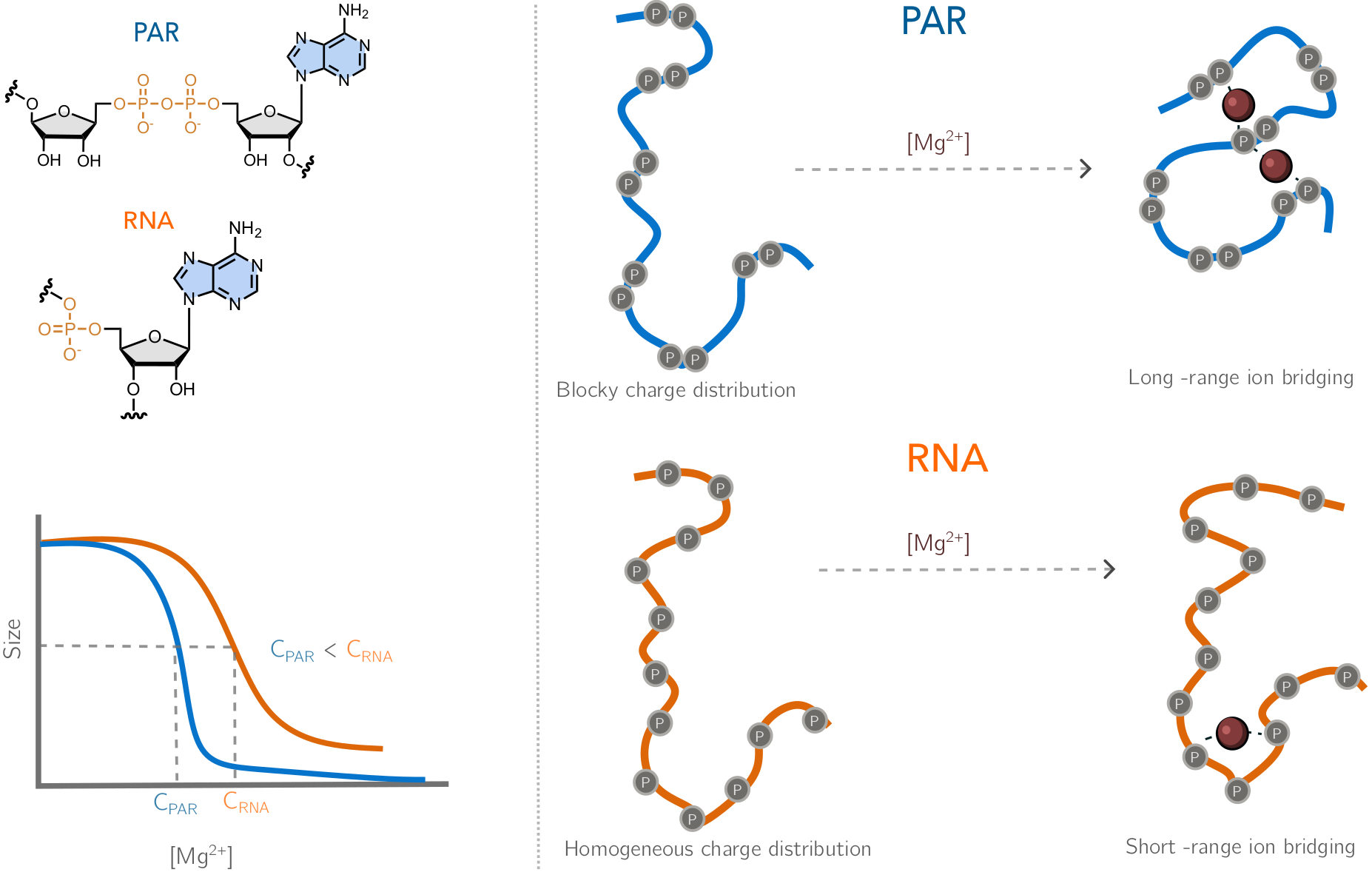 Poly(ADP-ribose) (PAR) exhibits ion-dependent structural properties distinct from RNALipika Baidya, Heyang Zhang, and Hung T NguyenNucleic Acids Res., 2026
Poly(ADP-ribose) (PAR) exhibits ion-dependent structural properties distinct from RNALipika Baidya, Heyang Zhang, and Hung T NguyenNucleic Acids Res., 2026Poly(ADP-ribose) (PAR) is a highly charged, intrinsically flexible nucleic-acid-like homopolymer composed of ADP-ribose units. It serves as a critical post-translational modification that is rapidly synthesized in response to DNA damage and participates in diverse important cellular processes, including DNA repair, chromatin remodeling, and RNA biogenesis, often by promoting biomolecular phase separation. However, PAR’s conformational heterogeneity coupled with limited experimental characterization has hindered a mechanistic understanding of these processes. To address this challenge, we develop a five-bead coarse-grained model of PAR and perform molecular dynamics simulations to compare its conformational ensemble and ion atmosphere with those of RNA across diverse ion environments. Our simulations reveal that PAR undergoes a markedly stronger structural transition than RNA in response to divalent ions, driven by its preferential ion binding and effective ion-mediated bridging between phosphate groups. These interactions facilitate cooperative conformational changes and produce a compact, ion-rich atmosphere that is fundamentally distinct from RNA. Together, our work provides a physically grounded model for PAR, uncovers molecular features that differentiate PAR from RNA, and offers mechanistic insight into how PAR nucleates phase separation and organizes protein interactions at sites of DNA damage.
-
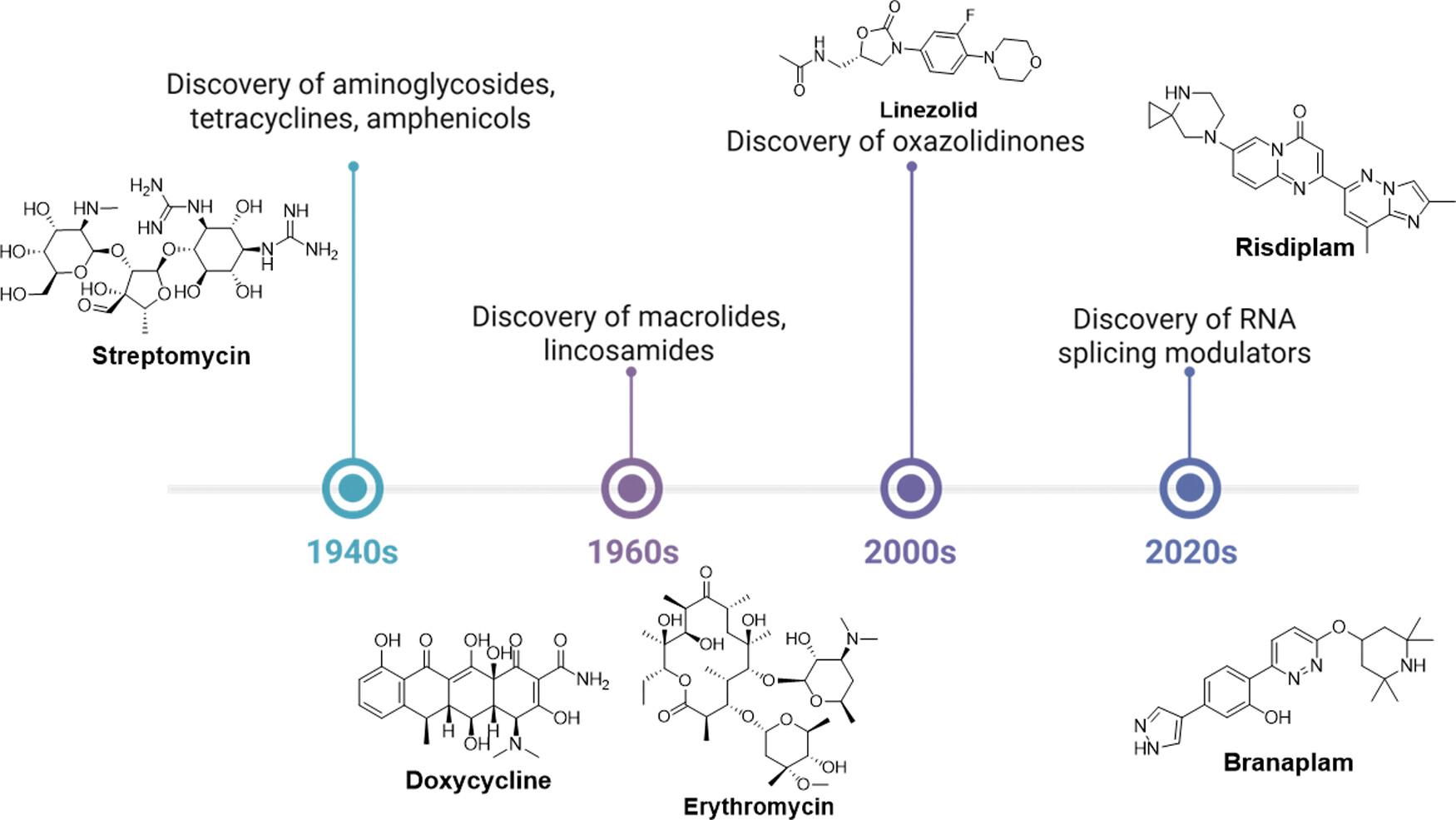 Small-Molecule Drug Discovery Targeting RNAs: Hope or Hype?Congbao Kang, HTN, David E. Heppner, Bin Yu, and Weijun XuJ. Med. Chem., 2026
Small-Molecule Drug Discovery Targeting RNAs: Hope or Hype?Congbao Kang, HTN, David E. Heppner, Bin Yu, and Weijun XuJ. Med. Chem., 2026


2025
-
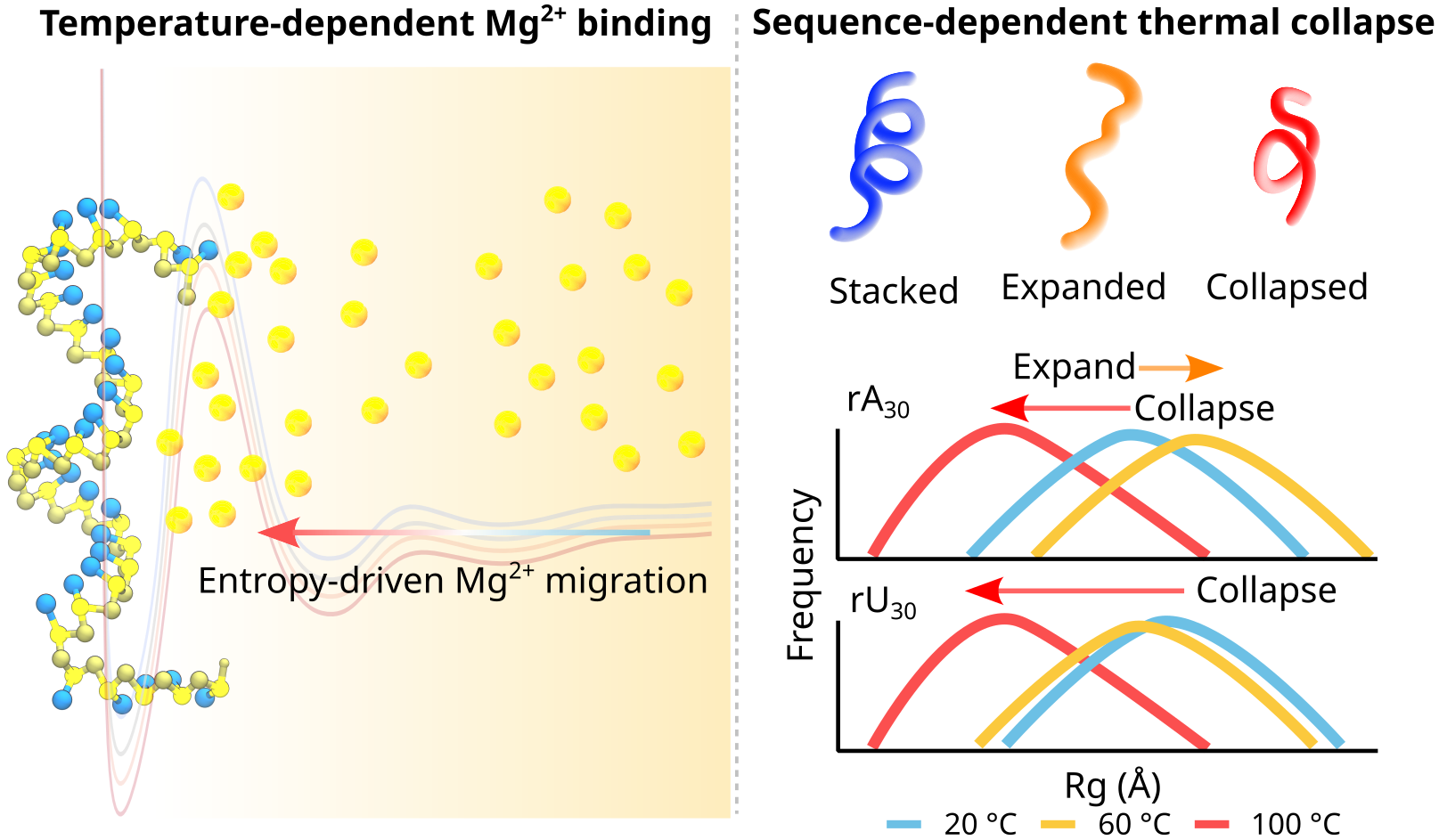 Temperature-dependent Ion Migration Underlies Sequence-Specific RNA CollapseHeyang Zhang, Hiranmay Maity, and HTNbioRxiv, 2025
Temperature-dependent Ion Migration Underlies Sequence-Specific RNA CollapseHeyang Zhang, Hiranmay Maity, and HTNbioRxiv, 2025Ions and temperature jointly regulate RNA structure, dynamics and phase behavior, yet their coupled effects remain poorly understood at the molecular level. Single-stranded RNA (ssRNA), a ubiquitous and functionally versatile class of RNA, presents a particularly challenging target due to its intrinsic flexibility and pronounced sensitivity to ionic and thermal perturbations. Here, we develop and apply coarse-grained simulations incorporating a temperature-dependent Mg2+-phosphate potential to elucidate how electrostatics, stacking, and hydration collectively determine ssRNA behavior. Our simulations quantitatively reproduce experimental SAXS profiles across a broad range of ionic conditions and reveal a non-monotonic temperature dependence of RNA compaction: ssRNAs expand upon heating, reach a sequence-specific maximum size, and then collapse as enhanced counterion condensation dominates. Rising temperature strengthens ion-RNA interactions, leading to a reorganization from diffusive to inner-sphere coordination, directly linking RNA collapse to ion dehydration. Our results establish that the ion atmosphere is a dynamic, sequence-encoded extension of RNA structure. This framework provides molecular insight into how temperature and ions govern RNA conformational transitions, offering a microscopic basis for RNA thermoadaptation, cold-induced misfolding, and RNA phase transitions.
-
 Controlling intermolecular base pairing in Drosophila germ granules by mRNA folding and its implications in fly developmentSiran Tian, HTN, Ziqing Ye, Silvi Rouskin, D. Thirumalai, and Tatjana TrcekNat. Commun., 2025
Controlling intermolecular base pairing in Drosophila germ granules by mRNA folding and its implications in fly developmentSiran Tian, HTN, Ziqing Ye, Silvi Rouskin, D. Thirumalai, and Tatjana TrcekNat. Commun., 2025Drosophila germ granules are enriched with mRNAs critical for development. Within them, mRNAs cluster through intermolecular interactions that may involve base pairing. Here we apply in silico, in vitro and in vivo approaches to examine the type and prevalence of these interactions. We show that RNA clustering can occur without extended sequence complementarity (stretches of six or more continuous complementary bases) and that mRNAs display similar level of foldedness within germ granules as outside. Our simulations predict that clustering is driven by scattered, surface-exposed bases, enabling intermolecular base pairing. Notably, engineered germ granule mRNAs containing exposed GC-rich complementary sequences within stem loops located in the 3′ untranslated region promote intermolecular interactions. However, these mRNAs are also expressed at lower levels, leading to developmental defects. Although germ granule mRNAs contain numerous GC-rich complementary sequences, RNA folding renders them inaccessible for intermolecular base pairing. We propose that RNA folding restricts intermolecular base pairing to maintain proper mRNA function within germ granules.
-
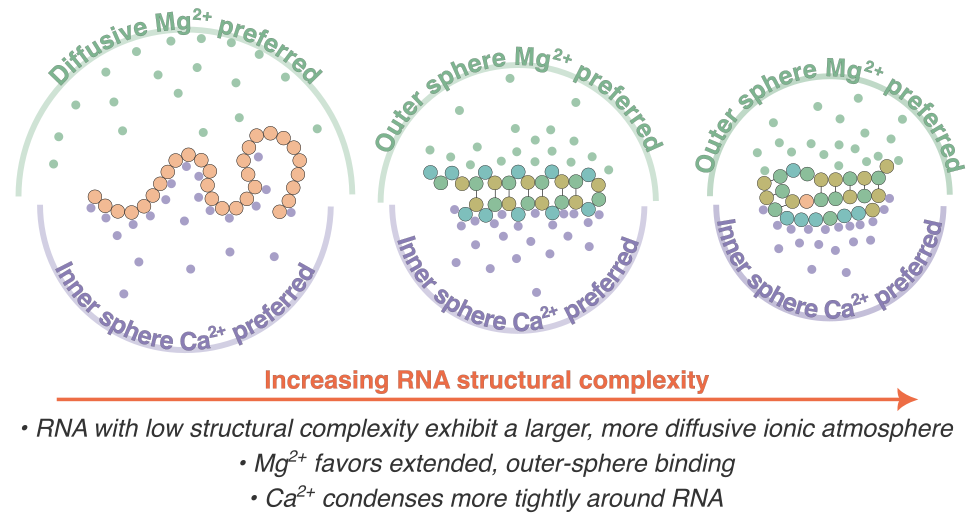 RNA Structural Complexity Dictates Its Ion AtmosphereHiranmay Maity, Heyang Zhang, D. Thirumalai, and HTNJ. Phys. Chem. Lett., 2025
RNA Structural Complexity Dictates Its Ion AtmosphereHiranmay Maity, Heyang Zhang, D. Thirumalai, and HTNJ. Phys. Chem. Lett., 2025Electrostatic interactions mediated by surrounding ions critically influence RNA behavior, yet flexible RNAs remain underexplored. We performed molecular dynamics simulations to examine three RNAs across the structural continuum: unstructured poly uridylic tract (rU30), a semiflexible cytosine-adenine-guanine (CAG) repeat, and a tightly folded pseudoknot. Despite similar net charges, rU30 attracts a diffuse Mg2+ cloud extending beyond two hydration shells, while the CAG repeat and pseudoknot favor more compact outer-sphere Mg2+ binding. In contrast, Ca2+ consistently forms inner-sphere contacts across all three RNAs. Remarkably, the ion atmospheres around unstructured RNAs extend farther into solution than that of the folded RNAs, significantly broadening their electrostatic sphere of influence. Nonetheless, the ion exchange kinetics remain virtually unchanged, demonstrating a surprising decoupling between spatial distribution and dynamical turnover. Our findings reveal RNA structural flexibility as a powerful lever for tuning ionic screening, with important implications for biomolecular recognition, phase separation, and biophysical properties of RNA-rich condensates.
-
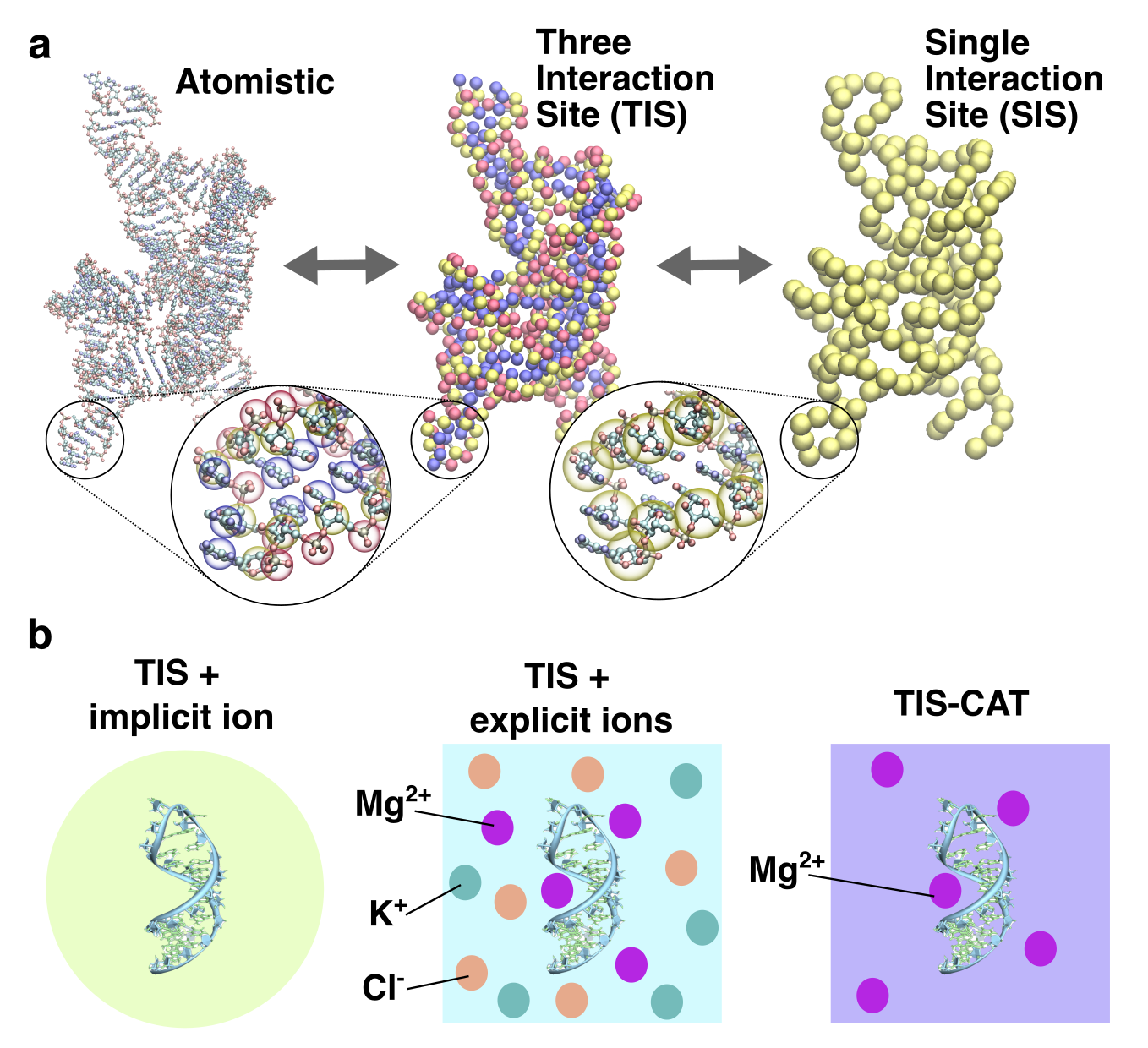 Minimal models for RNA simulationsD. Thirumalai, Naoto Hori, and HTNCurr. Opin. Struct. Biol., 2025
Minimal models for RNA simulationsD. Thirumalai, Naoto Hori, and HTNCurr. Opin. Struct. Biol., 2025The increasing importance of RNA as a prime player in biology can hardly be overstated. Problems in RNA, such as folding and RNA–RNA interactions that drive phase separation, require cations. Because experiments alone cannot reveal the dynamics of cation-RNA interactions, well calibrated theory and computations are needed to predict how ions control the behavior of RNA. The perspective describes the development and use of coarse-grained models at different resolutions. We focus on single- and three-interaction site models, in which electrostatic interactions are treated using a combination of explicit and implicit representations. Applications to the folding of ribozymes and riboswitches are discussed, with emphasis on the role of monovalent and divalent cations. We also discuss phase separation in low-complexity sequences. Challenges in the simulation of complex problems such as ribosome assembly and RNA chaperones, requiring developments of models for RNA-protein interactions, are pointed out.
-
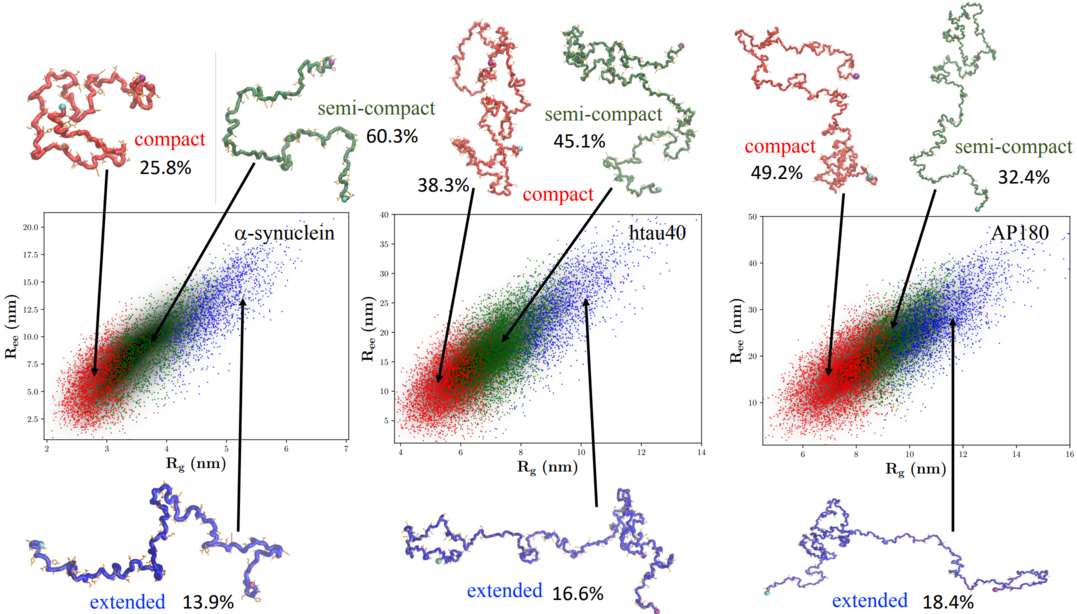 Sizes, conformational fluctuations, and SAXS profiles for intrinsically disordered proteinsMauro L. Mugnai, Debayan Chakraborty, HTN, Farkhad Maksudov, Abhinaw Kumar, Wade Zeno, Jeanne C. Stachowiak, John E. Straub, and 1 more authorProt. Sci., 2025
Sizes, conformational fluctuations, and SAXS profiles for intrinsically disordered proteinsMauro L. Mugnai, Debayan Chakraborty, HTN, Farkhad Maksudov, Abhinaw Kumar, Wade Zeno, Jeanne C. Stachowiak, John E. Straub, and 1 more authorProt. Sci., 2025The preponderance of intrinsically disordered proteins (IDPs) in the eukaryotic proteome, and their ability to interact with each other, and with folded proteins, RNA, and DNA for functional purposes, have made it important to quantitatively characterize their biophysical properties. Toward this end, we developed the transferable self-organized polymer (SOP-IDP) model to calculate the properties of several IDPs. The values of the radius of gyration (Rg) obtained from SOP-IDP simulations are in excellent agreement (correlation coefficient of 0.96) with those estimated from SAXS experiments. For AP180 and Epsin, the predicted values of the hydrodynamic radii (Rhs) are in nearly quantitative agreement with those from fluorescence correlation spectroscopy (FCS) experiments. Strikingly, the calculated SAXS profiles for 36 IDPs are also nearly superimposable on the experimental profiles. The dependence of Rg and the mean end-to-end distance (Ree) on chain length, N, follows Flory’s scaling law, Rα≈aαN^0.588 (α=g and ee), suggesting that globally IDPs behave as synthetic polymers in a good solvent. This finding depends on the solvent quality, which can be altered by changing variables such as pH and salt concentration. The values of ag and ae are 0.20 and 0.48 nm, respectively. Surprisingly, finite size corrections to scaling, expected on theoretical grounds, are negligible for Rg and Ree. In contrast, only by accounting for the finite sizes of the IDPs, the dependence of experimentally measurable Rh on N can be quantitatively explained using ν=0.588. Although Flory scaling law captures the estimates for Rg, Ree, and Rh accurately, the spread of the simulated data around the theoretical curve is suggestive of of sequence-specific features that emerge through a fine-grained analysis of the conformational ensembles using hierarchical clustering. Typically, the ensemble of conformations partitions into three distinct clusters, having different equilibrium populations and structural properties. Without any further readjustments to the parameters of the SOP-IDP model, we also obtained nearly quantitative agreement with paramagnetic relaxation enhancement (PRE) measurements for α-synuclein. The transferable SOP-IDP model sets the stage for several applications, including the study of phase separation in IDPs and interactions with nucleic acids.





2024
-
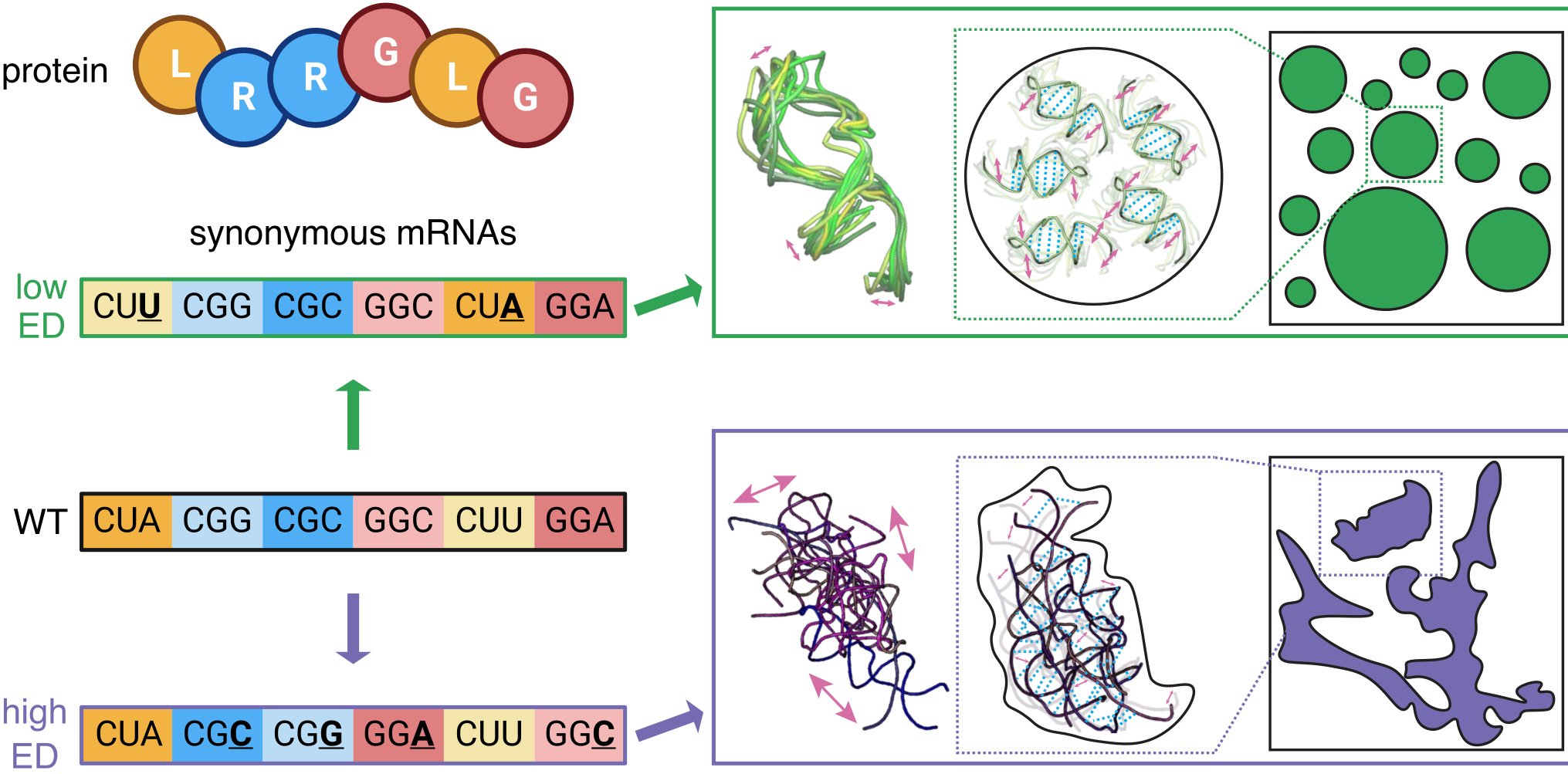 RNA encodes physical informationIan Seim, Vita Zhang, Ameya P. Jalihal, Benjamin M. Stormo, Sierra J. Cole, Joanne Ekena, HTN, D. Thirumalai, and 1 more authorbioRxiv, 2024
RNA encodes physical informationIan Seim, Vita Zhang, Ameya P. Jalihal, Benjamin M. Stormo, Sierra J. Cole, Joanne Ekena, HTN, D. Thirumalai, and 1 more authorbioRxiv, 2024Most amino acids are encoded by multiple codons, making the genetic code degenerate. Synonymous mutations affect protein translation and folding, but their impact on RNA itself is often neglected. We developed a genetic algorithm that introduces synonymous mutations to control the diversity of structures sampled by an mRNA. The behavior of the designed mRNAs reveals a physical code layered in the genetic code. We find that mRNA conformational heterogeneity directs physical properties and functional outputs of RNA-protein complexes and biomolecular condensates. The role of structure and disorder of proteins in biomolecular condensates is well appreciated, but we find that RNA conformational heterogeneity is equally important. This feature of RNA enables both evolution and engineers to build cellular structures with specific material and responsive properties.
-
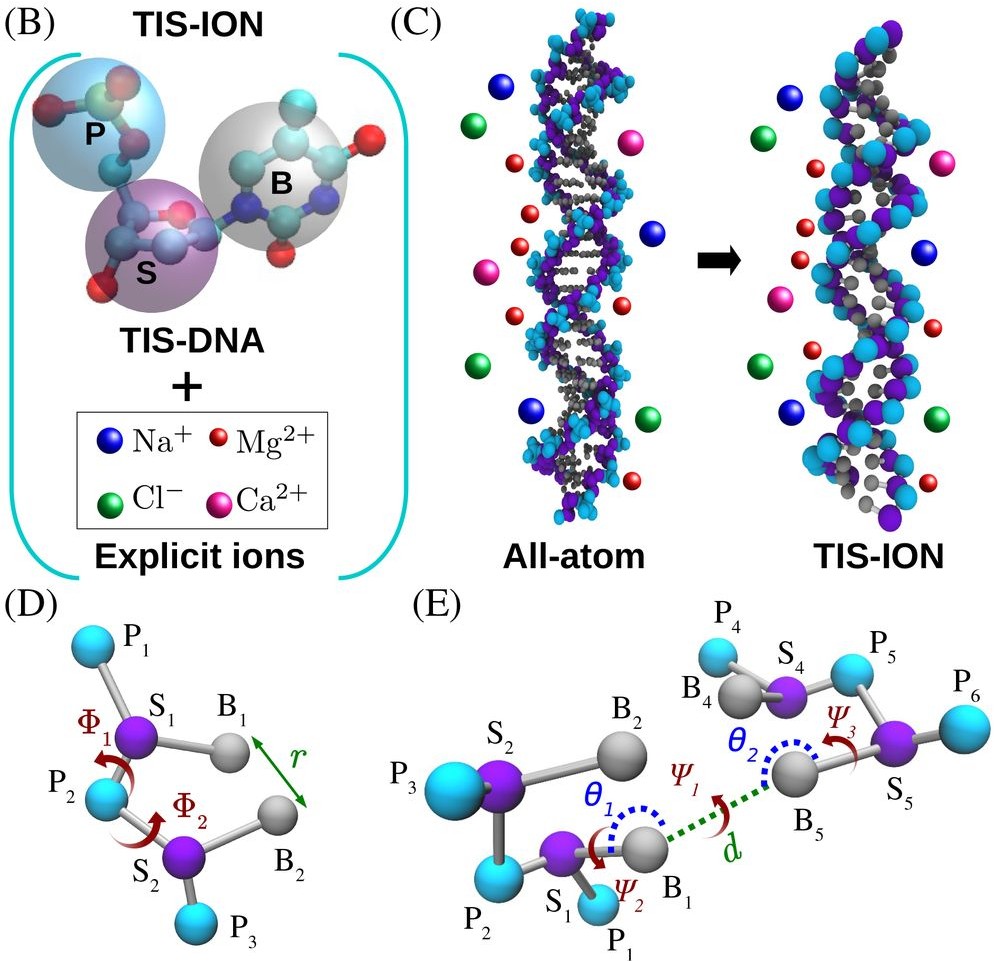 Competition between Stacking and Divalent Cation-Mediated Electrostatic Interactions Determines the Conformations of Short DNA SequencesBalaka Mondal, Debayan Chakraborty, Naoto Hori, HTN, and D. ThirumalaiJ. Chem. Theory Comput., 2024
Competition between Stacking and Divalent Cation-Mediated Electrostatic Interactions Determines the Conformations of Short DNA SequencesBalaka Mondal, Debayan Chakraborty, Naoto Hori, HTN, and D. ThirumalaiJ. Chem. Theory Comput., 2024Interplay between divalent cations (Mg2+ and Ca2+) and single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA), as well as stacking interactions, is important in nucleosome stability and phase separation in nucleic acids. Quantitative techniques accounting for ion–DNA interactions are needed to obtain insights into these and related problems. Toward this end, we created a sequence-dependent computational TIS-ION model that explicitly accounts for monovalent and divalent ions. Simulations of the rigid 24 base-pair (bp) dsDNA and flexible ssDNA sequences, dT30 and dA30, with varying amounts of the divalent cations show that the calculated excess number of ions around the dsDNA and ssDNA agree quantitatively with ion-counting experiments. Using an ensemble of all-atom structures generated from coarse-grained simulations, we calculated the small-angle X-ray scattering profiles, which are in excellent agreement with experiments. Although ion-counting experiments mask the differences between Mg2+ and Ca2+, we find that Mg2+ binds to the minor grooves and phosphate groups, whereas Ca2+ binds specifically to the minor groove. Both Mg2+ and Ca2+ exhibit a tendency to bind to the minor groove of DNA as opposed to the major groove. The dA30 conformations are dominated by stacking interactions, resulting in structures with considerable helical order. The near cancellation of the favorable stacking and unfavorable electrostatic interactions leads to dT30 populating an ensemble of heterogeneous conformations. The successful applications of the TIS-ION model are poised to confront many problems in DNA biophysics.
-
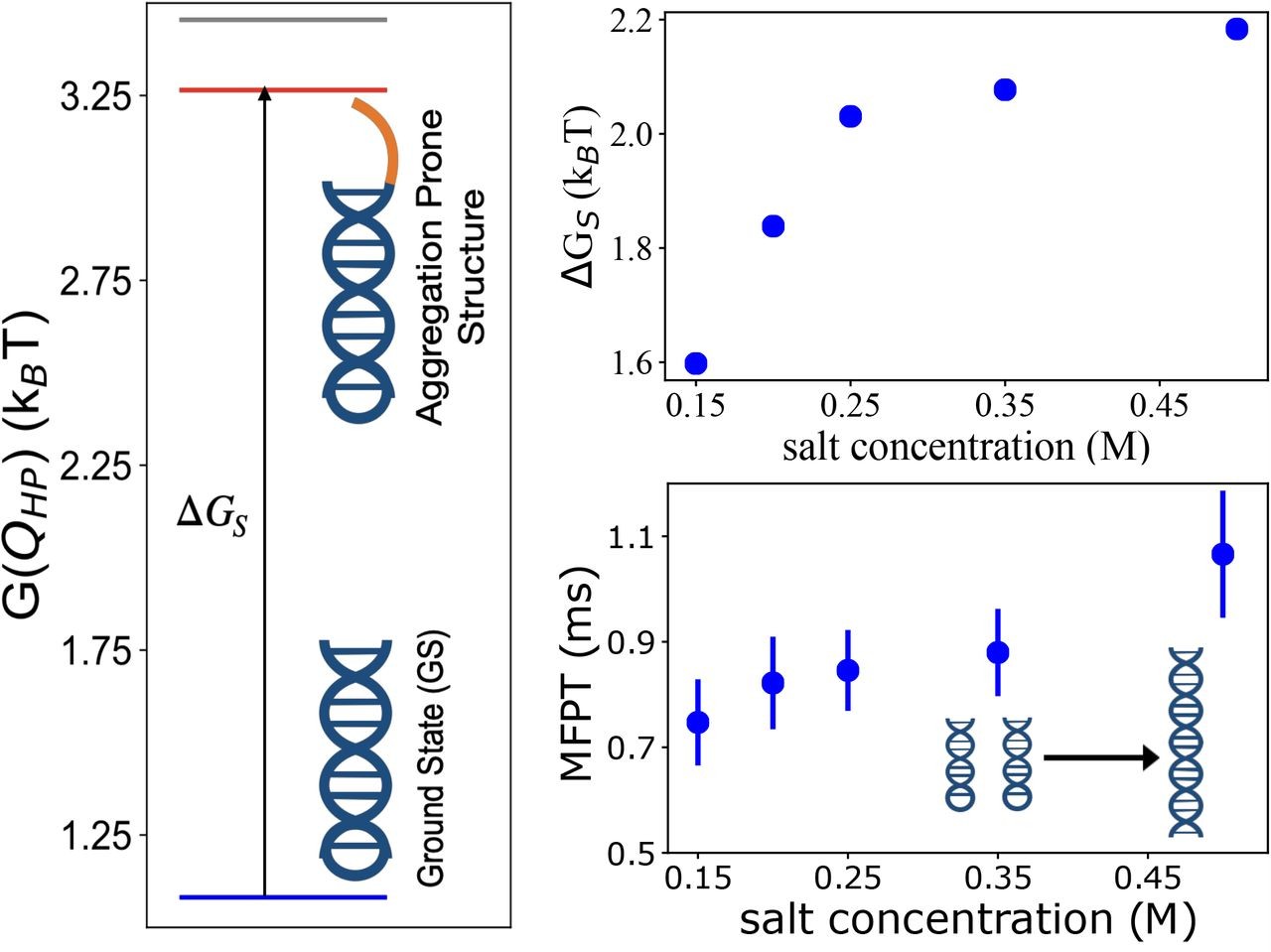 Salt-Dependent Self-Association of Trinucleotide Repeat RNA SequencesHiranmay Maity, HTN, Naoto Hori, and D. ThirumalaiJ. Phys. Chem. Lett., 2024
Salt-Dependent Self-Association of Trinucleotide Repeat RNA SequencesHiranmay Maity, HTN, Naoto Hori, and D. ThirumalaiJ. Phys. Chem. Lett., 2024Repeat RNA sequences self-associate to form condensates. Simulations of a coarse-grained single-interaction site model for (CAG)n (n = 30 and 31) show that the salt-dependent free energy gap, ΔGS, between the ground (perfect hairpin) and the excited state (slipped hairpin (SH) with one CAG overhang) of the monomer for (n even) is the primary factor that determines the rates and yield of self-assembly. For odd n, the free energy (GS) of the ground state, which is an SH, is used to predict the self-association kinetics. As the monovalent salt concentration, CS, increases, ΔGS and GS increase, which decreases the rates of dimer formation. In contrast, ΔGS for shuffled sequences, with the same length and sequence composition as (CAG)31, is larger, which suppresses their propensities to aggregate. Although demonstrated explicitly for (CAG) polymers, the finding of inverse correlation between the free energy gap and RNA aggregation is general.



2023
-
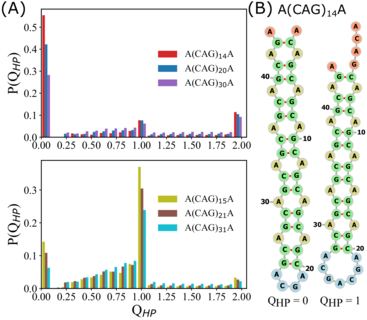 Odd-even disparity in the population of slipped hairpins in RNA repeat sequences with implications for phase separationHiranmay Maity, HTN, Naoto Hori, and D. ThirumalaiProc. Nat. Acad. Sci., 2023
Odd-even disparity in the population of slipped hairpins in RNA repeat sequences with implications for phase separationHiranmay Maity, HTN, Naoto Hori, and D. ThirumalaiProc. Nat. Acad. Sci., 2023Low-complexity nucleotide repeat sequences, which are implicated in several neurological disorders, undergo liquid–liquid phase separation (LLPS) provided the number of repeat units, n, exceeds a critical value. Here, we establish a link between the folding landscapes of the monomers of trinucleotide repeats and their propensity to self-associate. Simulations using a coarse-grained Self-Organized Polymer (SOP) model for (CAG)n repeats in monovalent salt solutions reproduce experimentally measured melting temperatures, which are available only for small n. By extending the simulations to large n, we show that the free-energy gap, ΔGS, between the ground state (GS) and slipped hairpin (SH) states is a predictor of aggregation propensity. The GS for even n is a perfect hairpin (PH), whereas it is a SH when n is odd. The value of ΔGS (zero for odd n) is larger for even n than for odd n. As a result, the rate of dimer formation is slower in (CAG)30 relative to (CAG)31, thus linking ΔGS to RNA–RNA association. The yield of the dimer decreases dramatically, compared to the wild type, in mutant sequences in which the population of the SH decreases substantially. Association between RNA chains is preceded by a transition to the SH even if the GS is a PH. The finding that the excitation spectrum—which depends on the exact sequence, n, and ionic conditions—is a predictor of self-association should also hold for other RNAs (mRNA for example) that undergo LLPS.

2022
-
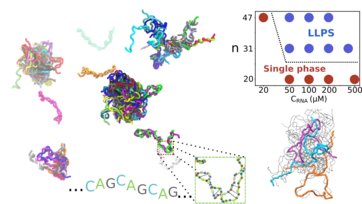 Condensates in RNA repeat sequences are heterogeneously organized and exhibit reptation dynamicsHTN, Naoto Hori, and D. ThirumalaiNat. Chem., 2022
Condensates in RNA repeat sequences are heterogeneously organized and exhibit reptation dynamicsHTN, Naoto Hori, and D. ThirumalaiNat. Chem., 2022Although it is known that RNA undergoes liquid–liquid phase separation, the interplay between the molecular driving forces and the emergent features of the condensates, such as their morphologies and dynamic properties, is not well understood. We introduce a coarse-grained model to simulate phase separation of trinucleotide repeat RNAs, which are implicated in neurological disorders. After establishing that the simulations reproduce key experimental findings, we show that once recruited inside the liquid droplets, the monomers transition from hairpin-like structures to extended states. Interactions between the monomers in the condensates result in the formation of an intricate and dense intermolecular network, which severely restrains the fluctuations and mobilities of the RNAs inside large droplets. In the largest densely packed high-viscosity droplets, the mobility of RNA chains is best characterized by reptation, reminiscent of the dynamics in polymer melts. Our work provides a microscopic framework for understanding liquid–liquid phase separation in RNA, which is not easily discernible in current experiments.

2020
-
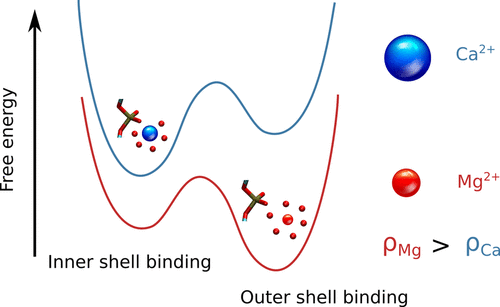 Charge Density of Cation Determines Inner versus Outer Shell Coordination to Phosphate in RNAHTN, and D. ThirumalaiJ. Phys. Chem. B, 2020
Charge Density of Cation Determines Inner versus Outer Shell Coordination to Phosphate in RNAHTN, and D. ThirumalaiJ. Phys. Chem. B, 2020Divalent cations are often required to fold RNA, which is a highly charged polyanion. Condensation of ions, such as Mg2+ or Ca2+, in the vicinity of RNA renormalizes the effective charges on the phosphate groups, thus minimizing the intra RNA electrostatic repulsion. The prevailing view is that divalent ions bind diffusively in a nonspecific manner. In sharp contrast, we arrive at the exact opposite conclusion using a theory for the interaction of ions with the phosphate groups using RISM theory in conjunction with simulations based on an accurate three-interaction-site RNA model. The divalent ions bind in a nucleotide-specific manner using either the inner (partially dehydrated) or outer (fully hydrated) shell coordination. The high charge density Mg2+ ion has a preference to bind to the outer shell, whereas the opposite is the case for Ca2+. Surprisingly, we find that bridging interactions, involving ions that are coordinated to two or more phosphate groups, play a crucial role in maintaining the integrity of the folded state. Their importance could become increasingly prominent as the size of the RNA increases. Because the modes of interaction of divalent ions with DNA are likely to be similar, we propose that specific inner and outer shell coordination could play a role in DNA condensation, and perhaps genome organization as well.

2019
-
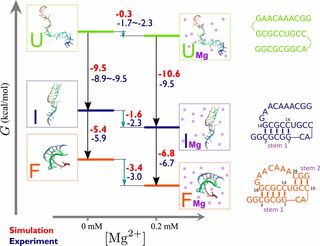 Theory and simulations for RNA folding in mixtures of monovalent and divalent cationsHTN, Naoto Hori, and D. ThirumalaiProc. Nat. Acad. Sci., 2019
Theory and simulations for RNA folding in mixtures of monovalent and divalent cationsHTN, Naoto Hori, and D. ThirumalaiProc. Nat. Acad. Sci., 2019RNA molecules cannot fold in the absence of counterions. Experiments are typically performed in the presence of monovalent and divalent cations. How to treat the impact of a solution containing a mixture of both ion types on RNA folding has remained a challenging problem for decades. By exploiting the large concentration difference between divalent and monovalent ions used in experiments, we develop a theory based on the reference interaction site model (RISM), which allows us to treat divalent cations explicitly while keeping the implicit screening effect due to monovalent ions. Our theory captures both the inner shell and outer shell coordination of divalent cations to phosphate groups, which we demonstrate is crucial for an accurate calculation of RNA folding thermodynamics. The RISM theory for ion–phosphate interactions when combined with simulations based on a transferable coarse-grained model allows us to predict accurately the folding of several RNA molecules in a mixture containing monovalent and divalent ions. The calculated folding free energies and ion-preferential coefficients for RNA molecules (pseudoknots, a fragment of the rRNA, and the aptamer domain of the adenine riboswitch) are in excellent agreement with experiments over a wide range of monovalent and divalent ion concentrations. Because the theory is general, it can be readily used to investigate ion and sequence effects on DNA properties.

2016
-
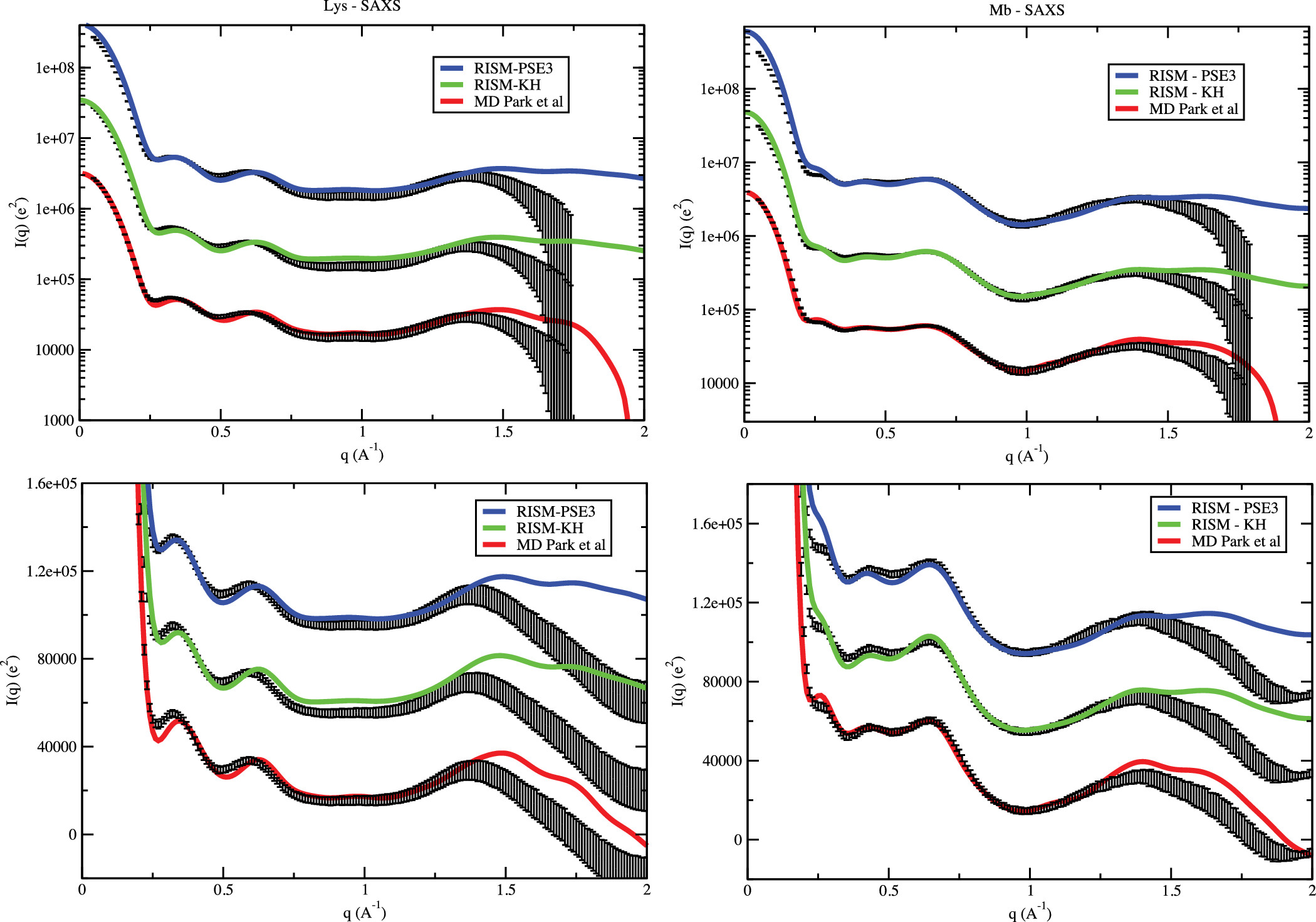
 Extracting water and ion distributions from solution x-ray scattering experimentsHTN, Suzette A. Pabit, Lois Pollack, and David A. CaseJ. Chem. Phys., 2016
Extracting water and ion distributions from solution x-ray scattering experimentsHTN, Suzette A. Pabit, Lois Pollack, and David A. CaseJ. Chem. Phys., 2016Small-angle X-ray scattering measurements can provide valuable information about the solvent environment around biomolecules, but it can be difficult to extract solvent-specific information from observed intensity profiles. Intensities are proportional to the square of scattering amplitudes, which are complex quantities. Amplitudes in the forward direction are real, and the contribution from a solute of known structure (and from the waters it excludes) can be estimated from theory; hence, the amplitude arising from the solvent environment can be computed by difference. We have found that this “square root subtraction scheme” can be extended to non-zero q values, out to 0.1 Å−1 for the systems considered here, since the phases arising from the solute and from the water environment are nearly identical in this angle range. This allows us to extract aspects of the water and ion distributions (beyond their total numbers), by combining experimental data for the complete system with calculations for the solutes. We use this approach to test molecular dynamics and integral-equation (3D-RISM (three-dimensional reference interaction site model)) models for solvent structure around myoglobin, lysozyme, and a 25 base-pair duplex DNA. Comparisons can be made both in Fourier space and in terms of the distribution of interatomic distances in real space. Generally, computed solvent distributions arising from the MD simulations fit experimental data better than those from 3D-RISM, even though the total small-angle X-ray scattering patterns are very similar; this illustrates the potential power of this sort of analysis to guide the development of computational models.

2014
-
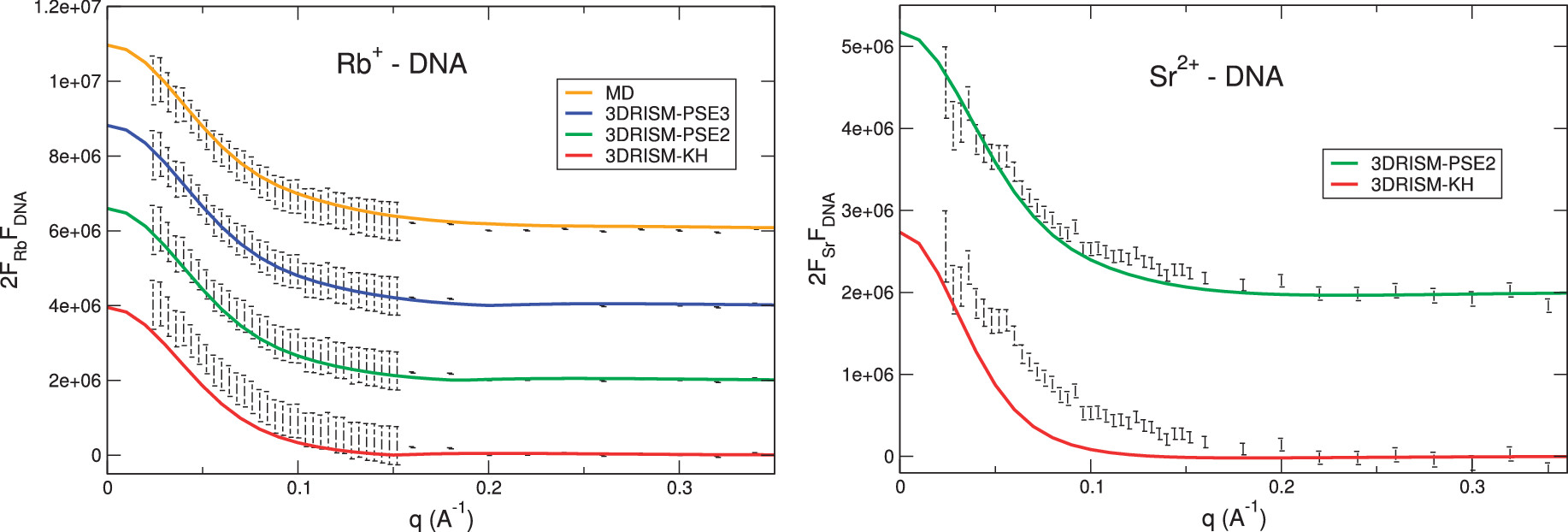
 Accurate small and wide angle x-ray scattering profiles from atomic models of proteins and nucleic acidsHTN, Suzette A. Pabit, Steve P. Meisburger, Lois Pollack, and David A. CaseJ. Chem. Phys., 2014
Accurate small and wide angle x-ray scattering profiles from atomic models of proteins and nucleic acidsHTN, Suzette A. Pabit, Steve P. Meisburger, Lois Pollack, and David A. CaseJ. Chem. Phys., 2014A new method is introduced to compute X-ray solution scattering profiles from atomic models of macromolecules. The three-dimensional version of the Reference Interaction Site Model (RISM) from liquid-state statistical mechanics is employed to compute the solvent distribution around the solute, including both water and ions. X-ray scattering profiles are computed from this distribution together with the solute geometry. We describe an efficient procedure for performing this calculation employing a Lebedev grid for the angular averaging. The intensity profiles (which involve no adjustable parameters) match experiment and molecular dynamics simulations up to wide angle for two proteins (lysozyme and myoglobin) in water, as well as the small-angle profiles for a dozen biomolecules taken from the BioIsis.net database. The RISM model is especially well-suited for studies of nucleic acids in salt solution. Use of fiber-diffraction models for the structure of duplex DNA in solution yields close agreement with the observed scattering profiles in both the small and wide angle scattering (SAXS and WAXS) regimes. In addition, computed profiles of anomalous SAXS signals (for Rb+ and Sr2+) emphasize the ionic contribution to scattering and are in reasonable agreement with experiment. In cases where an absolute calibration of the experimental data at q = 0 is available, one can extract a count of the excess number of waters and ions; computed values depend on the closure that is assumed in the solution of the Ornstein–Zernike equations, with results from the Kovalenko–Hirata closure being closest to experiment for the cases studied here.

2010
-
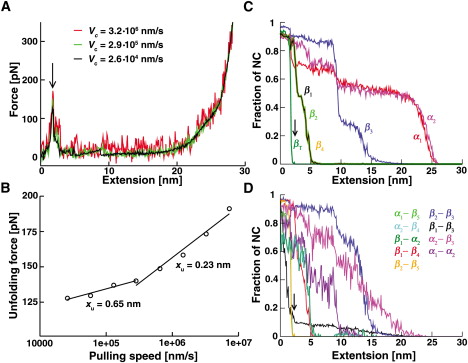 Mechanical Unfolding of Acylphosphatase Studied by Single-Molecule Force Spectroscopy and MD SimulationsGali Arad-Haase, Silvia G. Chuartzman, Shlomi Dagan, Reinat Nevo, Maksim Kouza, Binh Khanh Mai, HTN, Mai Suan Li, and 1 more authorBiophys. J., 2010
Mechanical Unfolding of Acylphosphatase Studied by Single-Molecule Force Spectroscopy and MD SimulationsGali Arad-Haase, Silvia G. Chuartzman, Shlomi Dagan, Reinat Nevo, Maksim Kouza, Binh Khanh Mai, HTN, Mai Suan Li, and 1 more authorBiophys. J., 2010Single-molecule manipulation methods provide a powerful means to study protein transitions. Here we combined single-molecule force spectroscopy and steered molecular-dynamics simulations to study the mechanical properties and unfolding behavior of the small enzyme acylphosphatase (AcP). We find that mechanical unfolding of AcP occurs at relatively low forces in an all-or-none fashion and is decelerated in the presence of a ligand, as observed in solution measurements. The prominent energy barrier for the transition is separated from the native state by a distance that is unusually long for α/β proteins. Unfolding is initiated at the C-terminal strand (βT) that lies at one edge of the β-sheet of AcP, followed by unraveling of the strand located at the other. The central strand of the sheet and the two helices in the protein unfold last. Ligand binding counteracts unfolding by stabilizing contacts between an arginine residue (Arg-23) and the catalytic loop, as well as with βT of AcP, which renders the force-bearing units of the protein resistant to force. This stabilizing effect may also account for the decelerated unfolding of ligand-bound AcP in the absence of force.

2009
- Top-hits for H1N1pdm identified by virtual screening using ensemble-based dockingHTN, Ly Le, and Thanh N. TruongPLoS currents, 2009
A list of 27 promising antiviral drugs is proposed for use against the H1N1pdm strain. Since the binding site of the H1N1pdm neuraminidase is similar to that of the bird flu H5N1, an effective means to quickly identify top candidates for use against H1N1pdm is to use known bird-flu drugs and the 27 compounds from the NCI diversity set which bind best to H5N1 neuraminidase. These compounds serve as viable candidates for docking against the H1N1pdm neuraminidase, using ensembles extracted from molecular dynamics simulations of the H1N1pdm system. The ranking order of these top candidates was found to be different from the previously published results for H5N1. The results indicated that the Oseltamivir (Tamiflu) and Peramivir drugs have higher ranking than Zanamivir (Relenza). However, six drug candidates were found to bind more effectively to H1N1pdm neuraminidase than Tamiflu. Detailed hydrogen bond network analysis for these six candidates is also provided.